State-of-the art translational Bioinformatics, as practiced by CUBI, must employ a large and growing number of systems and services. Data must be importable from different sources, meta data must be captured and managed, data is processed with various specialized pipelines, stored in central systems, and delivered using different platforms. CUBI Toolkit is a command line application for the translational Bioinformatician at CUBI to glue all of these disparate parts together.
CUBI Toolkit in Context
The following figure shows the context in which the CUBI Toolkit was conceived and developed. CUBI uses systems for data delivery and access for end users along with separate software and systems for data processing. CUBI Toolkit is a command line application that provides a unifying command line interface for gluing everything together.
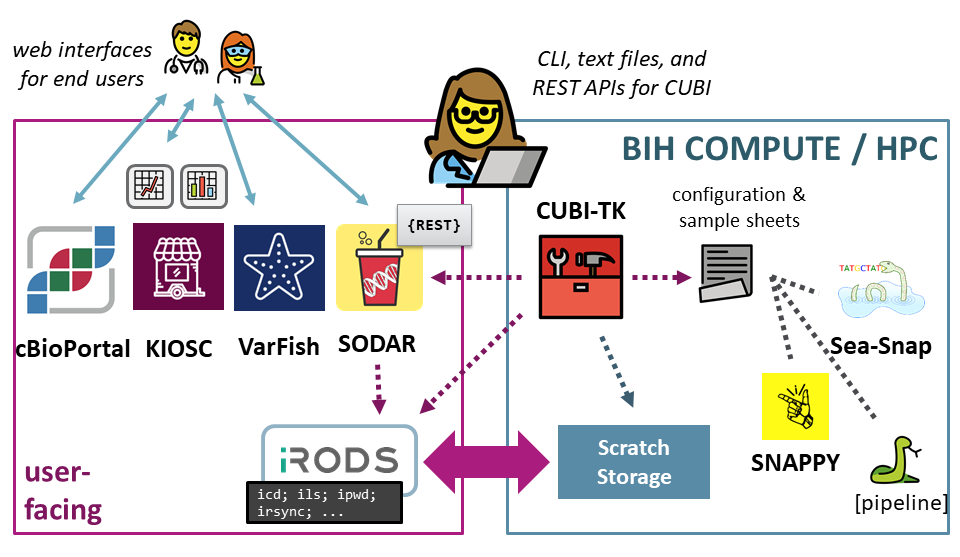
Illustration of CUBI Toolkit in context
Data may come from different sources that are available as folder structures on file systems. More and more data is initially uploaded into the SODAR system, however. CUBI Toolkit allows for the easy and robust ingestion of data into SODAR.
Generally, data is processed on the BIH High Performance Compute system. CUBI Toolkit facilitates the staging of meta data in the form of sample sheets and mass data files in the structure appropriate for the pipelines used by CUBI. Further, it makes running the pipelines easy and helps the user checking for the current pipeline running state.
After processing is complete, CUBI Toolkit supports the user by simplifying data upload into the iRODS backend of SODAR but also updating the meta data in SODAR from pipeline results. It also supports the user in uploading the pipeline results into platforms such as VarFish and cBioPortal.
CUBI Toolkit Principles
As all of CUBI data management software, CUBI Toolkit is leaning on ideas derived from the Unix Philosophy.
- Eschew monolithic systems, rather:
- start by providing sensibly small data types and objects and primitives to operate on them.
- use these primitives to construct more complex data types and operations, and
- based on this, hierarchically construct larger and more powerful system.
Each constructed layer that is available to the user on the command line is also available as a Python library. This facilitates using the tools in other software. One example of this is how the VarFish CLI Client is used as a library in CUBI Toolkit to provide the data types and primitives for interfacing with VarFish.
Germline Exome Analysis Use Case
One major example of how CUBI Toolkit supports Bioinformaticians in their daily work is in the Germline Exome Use Case. This is depicted in the following figure.
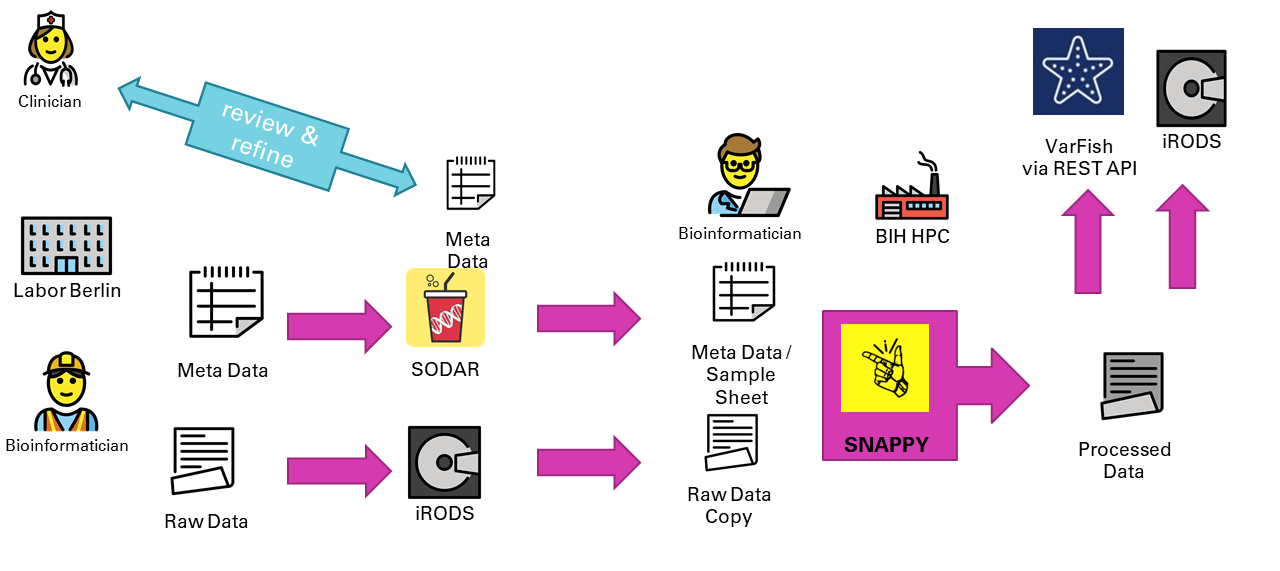
The Germline Exome Analysis Use Case
Sequencing data originates in an external sequencing lab. On completion, the bioinformatician uploads the raw sequencing data into SODAR and updates the sample sheet with the minimal information that is necessary for the data analysis from a lab-internal database. Both tasks are supported by CUBI Toolkit. Meta data is reviewed and curated by clinicians, physicians, and other data analysts by the lab.
Data and meta data is then staged to the HPC system where the data processing is performed by CUBI bioinformaticians. CUBI Toolkit facilitates in running the pipeline, verifying the results, and finally uploading the data back into SODAR for data access by the data analysts. Finally, annotated data from the pipeline is uploaded into the VarFish instance where it is available for analysis - again by the data analysists.
Related Software
- VarFish - VarFish, collaborative variant filtration
- SODAR - SODAR, the System for Omics Data Access and Retrieval
- SEA-SNAP, RNA-seq analysis with comprehensive reports (unpublished)
- SNAPPY Nucleic Acid Processing Pipeline (unpublished)
More Information
CUBI Contact
Attributions
Icons in the images are taken from OpenMoji.org.
Last modified: Mar 15, 2021